Research in the Young group focuses on the development and study of new materials for energy and electronics applications. We utilize a computational approach integrating materials informatics and machine learning, as well as first principles electronic structure methods such as density functional theory (DFT) and ab initio molecular dynamics (AIMD). A few of our active areas of research are summarized below.
HARNESSING 2D MATERIALS
Although Si-based technology has continually improved to keep pace with Moore’s law, the development of new atomically-thin planar (two dimensional or 2D) materials is a much more efficient way to meet the increasing need for the miniaturization of electronic components. Moreover, 2D materials are finding use in catalysis owing to their unique electronic structures, high surface areas, and relatively low cost. Because there is a vast array of structure types in 2D materials with chemical compositions further increasing the size of the phase space, this research direction focuses on using a high throughput computational screening approach to dramatically speed up the discovery process for these applications; this is combined with DFT and AIMD calculations to study the properties and stability of such materials. Additionally, investigating their interactions with other materials is critical for electronic device integration, while determining how to enhance their catalytic properties through the use of external stimuli or surface modification is needed since most pristine 2D materials are chemically inert.
Specific projects include (1) the discovery, design, and investigation of intrinsically ferroelectric and piezoelectric 2D materials and (2) understanding how strain, defects, atomic substitution, and chemical functionalization can be used to control these properties.
Representative Publications
Effect of chemical substitution and external strain on phase stability and ferroelectricity in two dimensional M2CT2 MXenes, M. Li, O. Omisakin, J. Young, Nanoscale 14 6970 (2022)
Valley Phenomena in the Candidate Phase Change Material WSe2(1-x)Te2x, S. M. Oliver, J. Young, S. Krylyuk, T. L. Reinecke, A. V. Davydov, P. M. Vora, Communications Physics 3 1 (2020)
Controlling the H to T’ Phase Transition in MoTe2 Through Chalcogen Doping, J. Young, T. L. Reinecke, Physical Chemistry Chemical Physics 19 31874 (2017)

Using density functional theory, we showed that the energy between two phases in MoTe2-based monolayers could be systematically tuned via chemical substitution
ELECTROLYTE ENGINEERING
The composition of an electrolyte must be specifically tailored to meet the needs of a given electrochemical application. In a battery, for example, critical parameters include electrochemical stability, formation of an adequate solid-electrolyte interphase (SEI) through decomposition, high cation diffusion, and safety; in catalysis, the makeup of the electrolyte can influence product selectivity and reduce overpotentials. However, the unimaginable amount of possible constituents and concentrations means that finding the optimal electrolyte composition can take many rounds of experimentation; moreover, many of the mechanisms underlying an electrolyte’s properties are not well understood. In this research direction, we aim to extend computational materials design beyond traditional solids to the design of liquid electrolytes. We use high throughput computational methods to screen potential electrolyte constituents (such as organic molecules, salts, and ionic liquid cations and anions) and link descriptors to desired properties, as well as a combination of density functional theory calculations and ab initio molecular dynamics to investigate the properties of electrolytes and gain an understanding of their interactions with surfaces. In this way we can quickly identify new electrolytes to meet important challenges in energy storage and catalysis.
Some representative projects in this research direction include (1) the study and design of electrolytes for high voltage batteries, (2) developing an understanding the role of ionic liquids in CO2 reduction, and (3) determining decomposition products of organic electrolytes and SEI formation on anodes.
Representative Publications
Ab initio determination of a simultaneous dual-ion charging mechanism for Ni0.25Mn0.75O2 through redox reactions of Ni 2+/Ni4+ and O2-/O, R. Shepard, S. Brennan, T. R. Juran, J. Young, M. Smeu, Journal of Materials Chemistry A 10 18916 (2022)
Computational Investigation of Enhanced Properties in Functionalized Carbon Nanotube Doped Polyvinyl Alcohol Gel Electrolyte Systems, E. S. Karaman, S. Mitra, J. Young, Physical Chemistry Chemical Physics 23 21286 (2021)
Preventing Electrolyte Decomposition on a Ca Metal Electrode Interface Using and Artificial Solid-Electrolyte Interphase, J. Young and M. Smeu, Advanced Theory and Simulation 4 2100018 (2021)
Comparative Study of Electrolyte Decomposition at Li, Ca, and Al Anode Interfaces, J. Young, T. Juran, P. Kulick, M. Smeu, ACS Applied Energy Materials 2 1676 (2019)
Ethylene Carbonate-Based Electrolyte Decomposition and Solid-Electrolyte Interphase Formation on Ca Metal Anodes, J. Young, M. Smeu, Journal of Physical Chemistry Letters 9 3295 (2018)
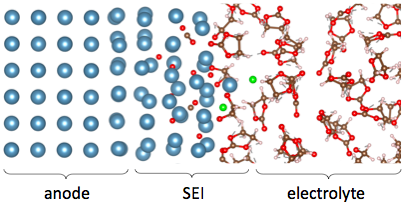
Schematic of a solid-electrolyte interphase forming between a metal anode and an electrolyte containing a salt dissolved in ethylene carbonate solvent. Generated using ab initio molecular dynamics.
MATERIALS FOR WATER TREATMENT
While the vast major of the Earth is covered by water (approximately 70%), freshwater suitable for human consumption constitutes only a small fraction of this; furthermore, much of this is polluted by compounds such as dyes, heavy metals, pharmaceuticals, and others. Treatment of polluted water will become increasingly important as the population and therefore water demand grows. The design of improved materials for water treatment is therefore an important topic, but requires an understanding of interfacial phenomena in aqueous environments. Adsorptive removal is a promising approach because of its high efficiency, simplicity, and low cost; to be a good adsorptive material a number of properties are needed, including thermal and chemical stability, high specific surface area, high selectivity, and regenerative capabilities. Another option is using catalysts to breakdown pollutants into other non-harmful species. Typically, metals like Pt or Pd are used to achieve this, but less expensive catalysts are desperately needed. In the Young group we use DFT and AIMD to design and understand such materials.
Representative Publications
Decoupling Electron- and Phase-Transfer Processes to Enhance Electrochemical Nitrate-to-Ammonia Conversion by Blending Hydrophobic PTFE Nanoparticles within the Electrocatalyst Layer, J. Gao, Q. Ma, J. Young, J. C. Crittenden, W. Zhang, Advanced Energy Materials (2022, accepted)
Elucidating facet dependent electronic and electrochemical properties of Cu2O nanocrystals using AFM/SCEM and DFT, Q. Ma, J. Young, S. Basuray, G. Cheng, J. Gao, N. Yao, W. Zhang, Nano Today 45 101538 (2022)
Effect of single atom Platinum (Pt) doping and facet dependent on the electronic structure and light absorption of Lanthanum Titanium Oxide (La2Ti2O7): A Density Functional Theory study, Q. Ma, W. Zhang, J. Young, Surface Science 715 121949 (2022)
APPLICATIONS AND DESIGN OF FERROELECTRICS
Ferroelectric materials, or those which display a spontaneous polarization that is switchable with an external electric field, have gained increasing interest for use in a variety of applications owing to this unique property. For example, ferroelectric materials have recently been proposed as a way to circumvent the Sabatier principle; because the direction of the polarization is switchable, the reactivity of the surface can be changed by applying an electric field, thereby dynamically changing its propensity to bind different intermediate species. This allows for the realization of notoriously challenging catalytic reactions. Furthermore, solar devices integrating ferroelectrics can overcome many of the limitations of traditional photovoltaics, as layers containing such materials can be used for both light absorption/carrier generation and charge separation since their internal electric field can directly drive charge separation. The overarching goal of this research direction is to overcome challenges in integrating ferroelectric materials in these applications both by discovering new ones and studying, understanding, and optimizing the properties of existing ones.
Specific projects include (1) developing an understanding of catalytic pathways and selectivity on ferroelectric surfaces, (2) studying how adsorption and reactions on a variety of 2D materials are affected as the polarization is switched and determining how catalytic reactions can be enhanced, and (3) discovering promising new low band gap ferroelectrics using a high throughput in silico approach and studying their properties using DFT.
Representative Publications
Inducing and Tuning Ferroelectricity via Octahedral Rotations, Cation Ordering, and Epitaxial Strain in Double Perovskite Iodide Superlattices, J. Young, J. M. Rondinelli, Physical Review Materials 2 065406 (2018)
Emergent Polar Phase in an Ultrashort-Period Brownmillerite Superlattice, J. Young, E. J. Moon, D. Mukherjee, V. Gopalan, N. Alem, S. J. May, J. M. Rondinelli, Journal of the American Chemical Society 139 2833 (2017)
Learning from Data to Design Functional Materials without Inversion Symmetry, P. V. Balachandran, J. Young, T. Lookman, J. M. Rondinelli, Nature Communications 8 14282 (2017)
Interplay of Cation Ordering and Ferroelectricity in Perovskite Tin Iodides: Designing Polar Halide Perovskites for Photovoltaic Applications, G. Gou, J. Young, X. Liu, J. M. Rondinelli, Inorganic Chemistry 56 26 (2017)
